
Services
Protein Identification Workflow

Our peptide and protein identification platform utilizes shotgun proteomic strategies to comprehensively identify the composition of user-provided protein samples. Samples are digested by trypsin (or other proteases), fractionated using liquid chromatography, and analyzed by tandem mass spectrometry. The chromatographic strategy employed is tailored to the complexity of the mixture and may include (1) 1D online reversed phase (RP) separations, (2) multidimensional online fractionation (MudPIT), and offline fractionation (typically SCX or high pH RP).
Quantitative Proteomics Workflow
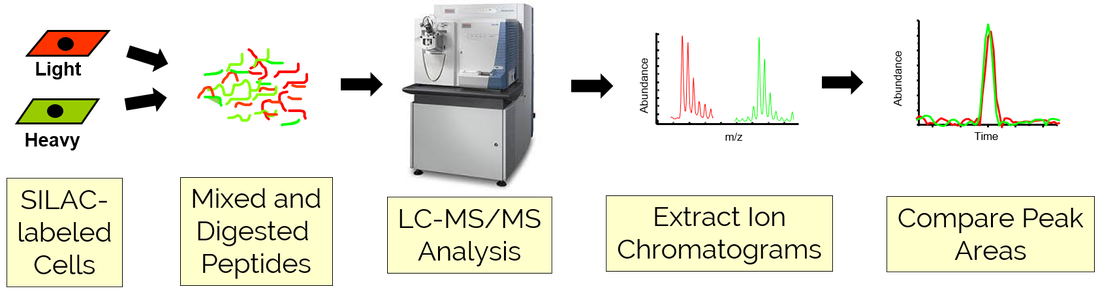
Our peptide and protein identification workflow is typically coupled to a quantitative proteomic strategy to enable the peptides and proteins identified in an experiment to be precisely and reliably compared across a set of samples. Available quantitative methods include (1) Stable Isotope Labeling of Amino Acid in Culture (SILAC), (2) stable isotope-encoded isobaric tagging (iTRAQ or TMT), and (3) label-free quantitation (LFQ) using either spectral counting or peptide intensity metrics. The choice of which quantitative technique to use is highly dependent on overall project goals and is typically decided in consultation with personnel from PRC.
PTM Analysis Workflow

A full-range of analytical pipelines for PTM enrichment, identification, and quantitation are available through the PRC. This includes both global and directed (i.e. focused on target of interest) PTM profiling and utilizes both chemical-based (e.g. titanium dioxide-based enrichment of phosphopeptides) and antibody-based (e.g. anti-acetylK or anti-diGly immunoaffinity isolation of acetylated and ubiquitinated peptides) enrichment techniques as necessary. These workflows are also coupled with different fractionation methods and quantitative strategies as necessary.
Targeted Proteomics Workflow
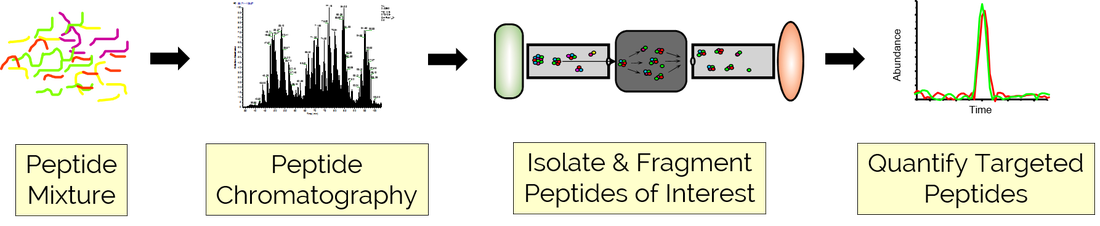
The UCLA PRC works with users to develop AQUA-type multiple reaction monitoring (MRM) or parallel reaction monitoring (PRM) assays that target a panel of biologically relevant peptides for rapid and robust quantitation and then apply those methods to analyze a variety of user-generated samples. These targeted workflows provide better quantitation and lower limits of detection as compared to unbiased discovery-based proteomic platforms and are well-suited for characterizing (1) the stoichiometry of complex protein assemblies, (2) the kinetics and activation state of different cellular signaling pathways, and (3) validation of disease biomarkers in a large cohort of clinical samples.